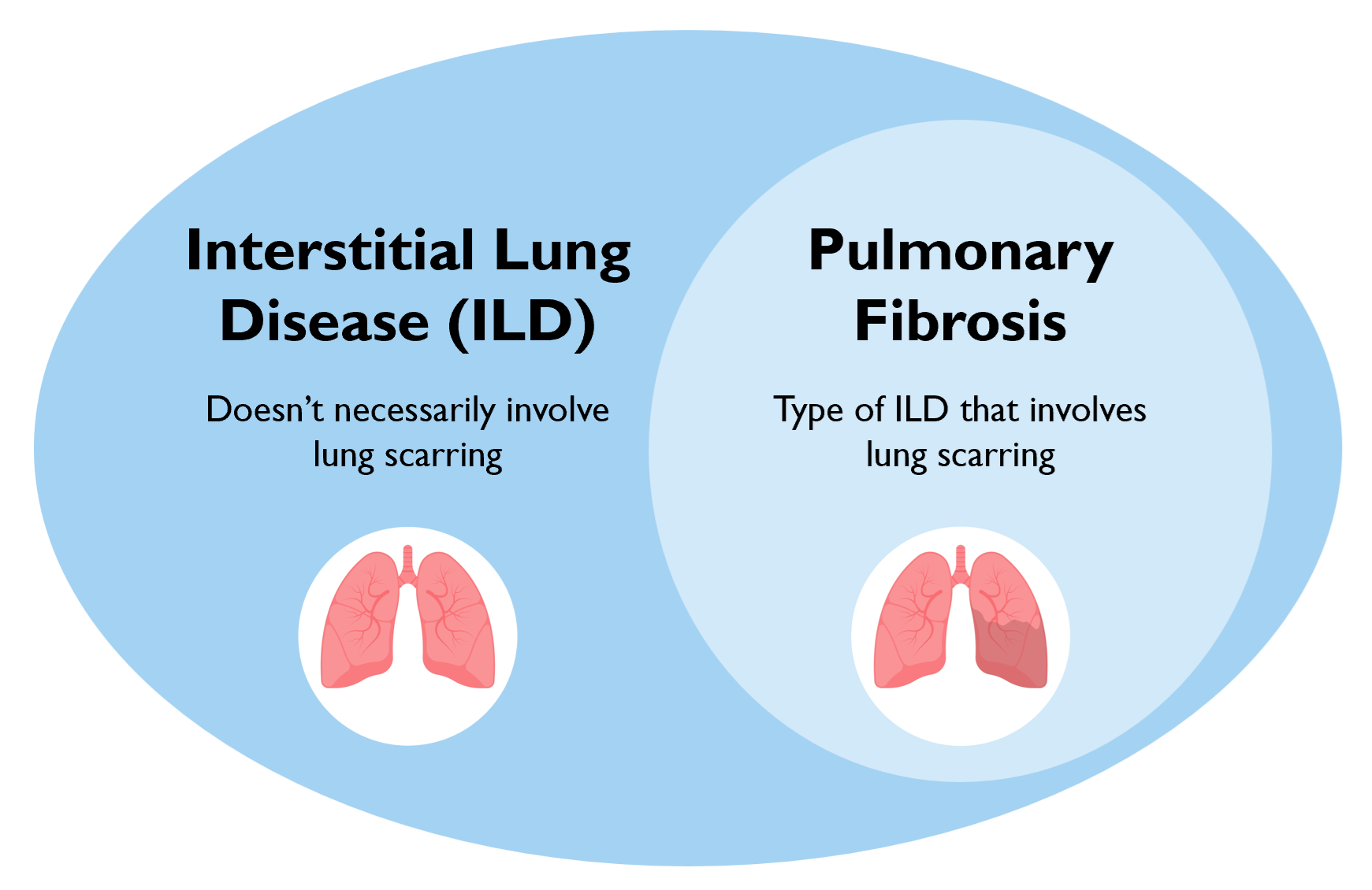
Interstitial lung diseases (ILDs) are a group of diseases that are characterized by chronic inflammation of the lung and may involve lung fibrosis or scarring.1 These diseases can manifest as inflammatory disease with little to no fibrosis, which are generally more responsive to anti-inflammatory and immunosuppressive medications. ILDs can also manifest as inflammatory disease with significant fibrosis, which generally has a poorer prognosis and limited response to therapies. There are also various manifestations of ILD in between these two extremes, characterized by inflammation and fibrosis to varying extents.
Pulmonary fibrosis occurs when lung parenchyma is replaced by scar tissue, and occurs in a subset of interstitial lung diseases.2 Pulmonary fibrosis can occur in various kinds of ILD that are caused by various triggers, including medications, autoimmune disease, and other allergens and exposures. When the cause of fibrosis is unknown, the disease is known as idiopathic pulmonary fibrosis (IPF).
Types of ILD
There are several broad subgroupings of ILDs:3,4
- Smoking-related, including respiratory bronchiolitis (RB), desquamative interstitial pneumonia (DIP), pulmonary Langerhans cell histiocytosis (PLCH), smoking-related interstitial fibrosis (SRIF), and combined pulmonary fibrosis and emphysema (CPFE)
- Exposure-related, including hypersensitivity pneumonitis and certain types of occupational lung disease
- Connective tissue disease-related, including from systemic lupus erythematosus (SLE), systemic sclerosis (SSc), Sjögren’s syndrome, rheumatoid arthritis (RA), mixed connective tissue disease (MCTD), systemic vasculitis, and anti-synthetase syndromes such as inflammatory myopathies, polymyositis (PM), and dermatomyositis (DM)
- Medication-induced, including from cardiovascular medications (e.g., amiodarone), antimicrobials (e.g., nitrofurantoin), biologics (e.g., tumor necrosis factor-inhibitors), neurologic medications (e.g., bromocriptine), herbal medications, and numerous anti-neoplastic medications
- Idiopathic pulmonary fibrosis (IPF)
- Pulmonary Sarcoidosis
- Lung disease with cysts or airspace filling
Every subtype of ILD may or may not include pulmonary fibrosis.4 Pulmonary fibrosis can be recognized based on characteristic findings on computed tomography (CT) images. Fibrosis can be recognized on high-resolution computed tomography (HRCT) through the presence of traction bronchiectasis and/or honeycombing.
ILD is rare, with the most common types including IPF (3-9 per 100,000 people)5, hypersensitivity pneumonitis (1.67-2.71 per 100,000 people)6, and connective tissue disease (prevalence varies by subtype).
References
- King TE, Jr. Clinical advances in the diagnosis and therapy of the interstitial lung diseases. Am J Respir Crit Care Med. Aug 1 2005;172(3):268-79. doi:10.1164/rccm.200503-483OE
- Murray L, Homer RJ, Gulati M, Herzog E. Pulmonary Fibrosis. In: McManus LM, Mitchell RN, eds. Pathobiology of Human Disease. Academic Press; 2014:2636-2653.
- Kalchiem-Dekel O, Galvin JR, Burke AP, Atamas SP, Todd NW. Interstitial Lung Disease and Pulmonary Fibrosis: A Practical Approach for General Medicine Physicians with Focus on the Medical History. J Clin Med. Nov 24 2018;7(12)doi:10.3390/jcm7120476
- Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. May 1 2022;205(9):e18-e47. doi:10.1164/rccm.202202-0399ST
- Maher TM, Bendstrup E, Dron L, et al. Global incidence and prevalence of idiopathic pulmonary fibrosis. Respir Res. Jul 7 2021;22(1):197. doi:10.1186/s12931-021-01791-z
- Fernández Pérez ER, Kong AM, Raimundo K, Koelsch TL, Kulkarni R, Cole AL. Epidemiology of Hypersensitivity Pneumonitis among an Insured Population in the United States: A Claims-based Cohort Analysis. Ann Am Thorac Soc. Apr 2018;15(4):460-469. doi:10.1513/AnnalsATS.201704-288OC